Measuring mRNA transcript abundance with RNASeq methods offers clinical researchers a powerful platform to examine changes in the expression levels of multiple biomarkers.
The power of targeted RNASeq lies in its ability to count the number of reads of a given transcript in a sample as a surrogate for expression level, much like a digital PCR system but able to measure 100s of biomarkers simultaneously.
A challenge to widespread adoption of RNASeq transcript abundance measurement for clinical studies and diagnostics is yield biases between targets affecting accuracy and reproducibility of the assay. Implementing a unique molecular index (UMI) approach can reduce bias, but at a cost of >8 fold increase in read depth.
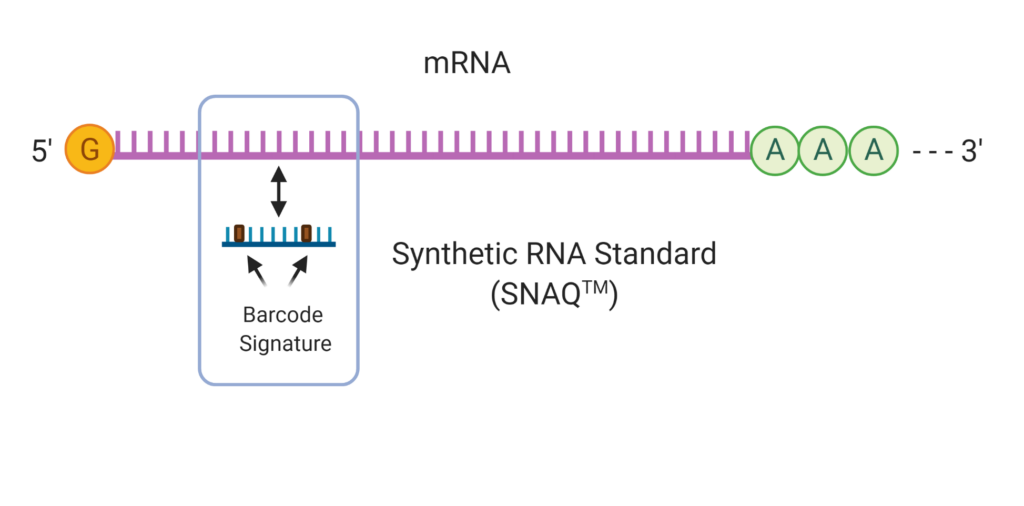
With SNAQ-TA, multiplexed spike-in RNA standards are used to standardize the measurement of transcripts by controlling for bias in measurements through the workflow. Specific standards matching target mRNA are created and spiked into the samples at the RT step. The ratio between target and standard are maintained during library prep and detection.
The result is near digital PCR-like accuracy across a greater dynamic range (up to 106 log) than dPCR methods. The ratio of standard to target concentration also permits the measure of high and low abundance targets in the same sample with significantly less reads, enabling increased sample throughput and lower per sample costs.
Additional Resources: